技术原理: |
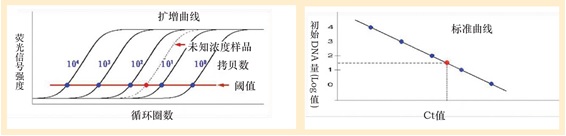 |
| Real Time PCR的定量原理图 |
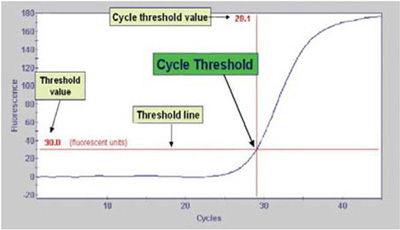 |
| 阈值和Ct值的相互关系图 |
| 决定系数(r Squared) 反映标准曲线的直线性,理想值应大于0.98,越接近于1说明直线性越好,定量越准确。 斜率(slope) 反映PCR扩增效率,-0.25~-0.33(根据不同装置数值不同)。 扩增效率(E) 理想扩增效率应在0.8﹤E﹤1.2。 |
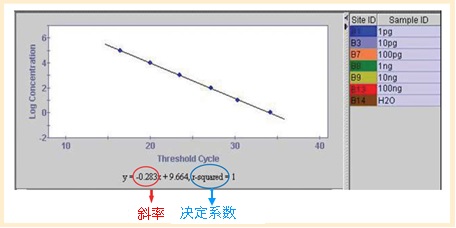 |
| 标准曲线的评价指标图例 |
| 应用进展: Real Time PCR是具有划时代意义的技术,具有操作简单、快速方便、灵敏度高、重复性好、污染率低等优点,不仅应用于基因表达解析,还广泛应用于SNP分型解析、物种鉴定、病毒和病原菌的检测、转基因食品的定量分析、导入基因拷贝数的解析等诸多领域。 阈值(threshold): 在扩增曲线的指数增长区域内适当位置上设定的荧光检出界限。 Ct值(Cycle threshold): 表示每个PCR反应管内荧光信号到达设定的域值时所经历的循环数。 经数学理论证明,Ct值与起始模板数的对数值成反比线性关系。 |
必须根据具体的实验用途进行选择。如果用于区别同源性高的序列以及进行SNP分型解析等多重PCR检测,荧光探针法无可替代,而进行除此之外的Real Time PCR实验,我们推荐使用简单易行、成本低的嵌合荧光嵌合法。 可以参考如下信息:
| 嵌合荧光法 | 探针法 | |
| 优点 | 简单易行,成本较低,无需合成特异性探针。 | 特异性强,能进行多重PCR。 |
| 缺点 | 对扩增的特异性要求高;不能进行多重PCR。 | 需要设计特异性探针,成本较高;有时探针设计困难。 |
探针法通常是使用5’端带有荧光物质(如:FAM等),3’端带有淬灭物质(如:TAMRA、BHQ等)的探针进行荧光检测的方法。当探针完整时,5’端的荧光物质受到3’端的淬灭物质的制约,不能检出荧光。探针的报告基团的发射波长要在淬灭基团的吸收波长范围内。
| 淬灭基团种类 | 淬灭基团性质 | 使用的报告基团种类 | |
| TAMRA | 荧光物质 |
FAM,HEX,TET,JOE等 | |
| BHQ系列染料 | BHQ1 | 非荧光物质 |
FAM,HEX,TET,JOE等 |
| BHQ2 | 非荧光物质 |
Cy3,TAMRA,ROX,Texas Red,Cy5等 | |
| BHQ3 | 非荧光物质 |
Cy5等 | |
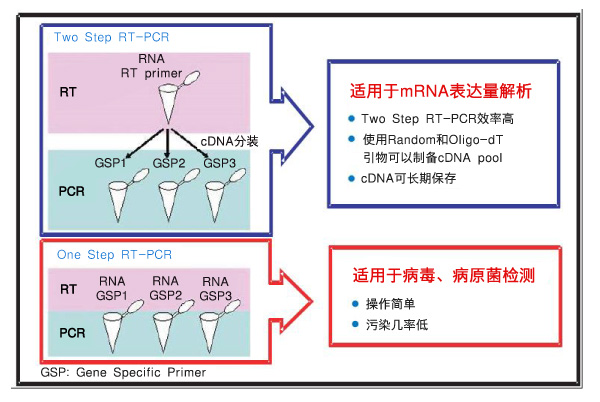
基因表达解析等绝大部分RT-PCR,非常适合选择Two Step RT-PCR,而RNA病毒检测等以基因检测为目的RT-PCR,应优先考虑防止污染,所以建议选择One Step RT-PCR。
1. 进行Two Step RT-PCR反应时,可选择Random Primer、Oligo dT Primer或基因的特异性引物进行反转录反应,然后再将一部分反转录反应液(cDNA)作为模板添加到Real Time PCR反应液中。使用反转录引物时可以根据实验目的选择合适的反转录引物,如果选择Random Primer或Oligo dT Primer,合成的cDNA可用于复数种类基因的检测,cDNA溶液可以稳定地长期保存,供以后解析使用。
2. One Step RT-PCR的反转录反应和PCR反应在同一反应管内连续进行,操作简单,污染几率低。但此时的反转录反应和PCR反应都并非为各自的适宜条件,所以One Step RT-PCR通常比Two Step RT-PCR反应效率低。此外,与Two Step RT-PCR反应相比,反转录酶等的使用量多,每个反应的成本相对较高。One Step RT-PCR反应的反转录反应引物只能使用基因的特异性PCR下游引物,而不能使用Random Primer或Oligo dT Primer共用引物。
绝对定量与相对定量有如下差别:
1. 绝对定量:是对未知样品的绝对量(拷贝数)进行测定的方法;通常应用于病毒、细菌、衣原体、支原体的定量检测及转基因食品的检测。
2. 相对定量:是分别测定目的基因和参比基因的量,再求出对于参比基因的目的基因的相对量,最后再进行样品间相对量的比较。主要用于检测细胞mRNA表达量的变化;比较不同组织的mRNA表达差异;验证基因芯片、siRNA干扰的实验结果。
荧光定量实验的成功因素如下:
1. 优良试剂的选择。
Real Time PCR试剂的选择是否合适,也是实验成功的关键点,所以在这里有必要对试剂的选择要点加以说明。
a. 标准曲线的质量直接影响定量的准确性。建议使用标准品专用稀释Buffer稀释标准品。
b. 根据采用的荧光检测方法进行试剂选择。选择探针法专用试剂或嵌合法专用试剂。
c. Real Time RT-PCR要根据实验目的进行试剂选择。mRNA表达量分析选择Two Step RT-PCR试剂,病毒病原菌检测选择One Step RT-PCR试剂。
d. Real Time PCR 反应通常样品数量较多,操作步骤过多易产生错误。最好选择PCR反应用Buffer、DNA聚合酶、dNTP等试剂已经预混在一起的2×Premix Type试剂,操作更加简单方便。同时,使用的试剂最好不需要在进行Mg2+浓度的优化等实验。
e. Real Time PCR反应对反应特异性要求高。最好选择应用Hot Start DNA聚合酶的试剂。但有一点需要强调,Hot Start DNA聚合酶有两种类型,即化学修饰型和抗体结合型。
使用这两种Hot Start DNA聚合酶的PCR反应条件不同,使用化学修饰型Hot Start DNA聚合酶,需要在PCR条件的最初步骤设置10~15分钟的热变性程序,以恢复DNA聚合酶的活性;而抗体结合型Hot Start DNA聚合酶,不需要长时间的热变性步骤,通常的PCR条件即可恢复DNA聚合酶的活性,最适合快速PCR反应。
f. 所选试剂要与使用的Real Time PCR检测系统相匹配。最好选择对各种机型装置都显示良好反应性能的试剂。
2. 引物和探针的设计。
引物设计原则
扩增片段大小 |
80-150 bp(尽量限制在300 bp以内) |
Primer长度 |
17-25 base |
GC含量 |
40-60%(最好45-55%) |
Tm值 |
两条引物的Tm值尽量接近,引用专用软件计算Tm值 |
序列 |
整体上碱基不能过偏,局部避免GC rich或AT rich(特别是3’端) |
3’末端序列 |
避免GC含量过高,3’末端碱基最好为G或C,尽量避免3’末端碱基为T |
互补性 |
引物内部或两条引物之间避免3 base以上的互补序列,引物3’末端避免2 base以上的互补序列 |
特异性 |
使用BLAST检索,确认引物特异性 |
RT-PCR用引物 |
尽量在Exon junction上设计引物,限制基因组DNA扩增 |
Probe长度 |
20-24 base |
Tm值 |
探针的Tm比引物高8-10℃ |
| 序列 | 在目的序列GC含量相对较高的区域设计,整体上碱基不能过偏,局部避免GC rich或AT rich(特别是3’端) |
5’末端序列 |
探针5’末端的第一个碱基不能是G |
互补性 |
Probe内部或Probe与两条引物之间避免3 base以上的互补序列 |
特异性 |
BLAST检索确认Probe特异性 |
3. 反应条件的优化
依据所使用的产品说明书进行反应条件的优化。
4. 实验操作
必须分区操作。通常分为:反应液调制区;模板添加/样品制备区;PCR反应区。
进行Real Time PCR首先要确认的工作如下:
1. 引物和探针的序列是否有错误?组合是否正确?
2. Total RNA是否有分解的可能?
3. 各种试剂是否忘记加入?
4. PCR条件和荧光检出步骤是否设定正确?
如果有成功反应经历的模板(Positive Control),设置Positive Control反应,很容易进行以上项目的确认。Total RNA一定要通过电泳和OD分析确认其质量。
有时Total RNA或cDNA中含有对反转录反应和PCR反应的阻害物质(如RNA提取时使用的有机溶剂等)。为了确认这样的阻害物质的有无,使用较高浓度的模板按3~4个梯度稀释,并使用其进行Real Time RT-PCR反应或Real Time PCR反应。如果无阻害物质存在,得到的Ct值会依存模板浓度的变化而变化;如果有阻害物质存在,就会发现高浓度的模板有反应性能下降的现象。
| 扩增曲线存在问题时,首先应确认基线校正或ROX校正前的原始曲线,扩增曲线的荧光信号值低多数是由于背景荧光信号值过高造成的。如下图所示,A的原始扩增曲线背景荧光信号值约为280,其基线校正后的扩增曲线显示较高的荧光信号值(约500);而B的原始扩增曲线背景荧光信号值可达800,其基线校正后的扩增曲线荧光信号值较低(约150)。使用嵌合荧光法进行检测时,背景荧光信号偏高大多数是由于模板量过高,染料嵌入到初始模板DNA中发出荧光造成的。使用荧光探针法进行检测时,背景荧光信号偏高大多是由于设计的探针质量差使淬灭基团的淬灭作用不充分、报告基团和淬灭基团的搭配不合理、探针的荧光标记效率低等原因造成的。 |
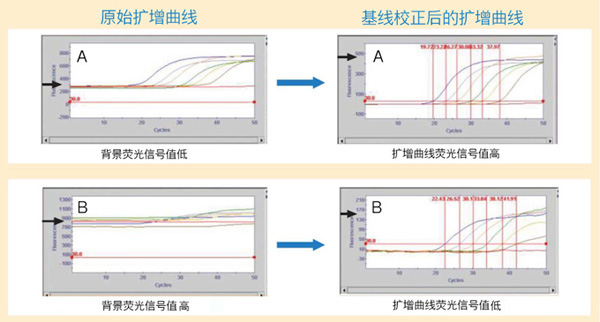 |
| 背景荧光信号值确认图例 |
出现这种现象一般有两种可能,如下图所示。 |
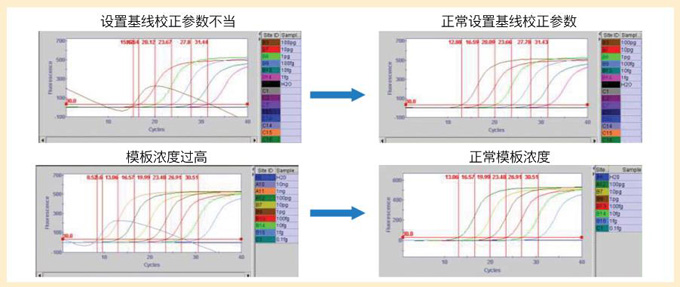 |
| 扩增曲线朝右下方下落的解决图例 |
由标准曲线的斜率计算得到的PCR扩增效率低时,可以考虑以下几点原因。
1. PCR反应性能差。引物、试剂、PCR条件的再摸索。
2. PCR阻害物质的混入。模板提取方法的再摸索。
3. 标准品稀释不准确。使用专用标准品稀释Buffer。
稀释的低浓度标准品常常有易分解不稳定的现象产生,所以使用的稀释液中最好加入与实验材料不同生物种的tRNA和rRNA以起保护作用。但偶尔也有因加入的tRNA和rRNA的序列与目的基因序列具有同源性而产生的干扰现象。如果使用不含tRNA和rRNA的标准品稀释专用试剂EASY Dilution(for Real Time PCR)(Code No. 9160),将不会有这方面的顾虑。
PCR扩增效率比理论值偏高,大多是由于反应体系中含有PCR阻害物质,需要改进模板提取方法。另外,使用嵌合荧光法检测时,有时非特异性扩增也会导致PCR扩增效率高,要通过融解曲线分析加以确认。
首先怀疑发生了目的片段以外的扩增。
|
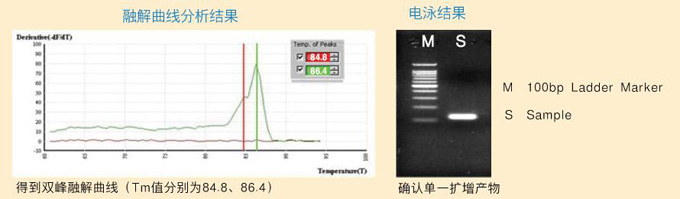 |
| 融解曲线分析特例 |
以前,GAPDH,β-actin基因常被作为管家基因使用,但近几年也常有这些基因因样品经药物等处理后表达量发生变化的报道。目前,很多研究者认为一种管家基因有可能校正不充分,同时使用两个或两个以上的复数管家基
因进行校正更值得信赖。当然这种校正方法也是非常耗费精力的,通常要选择若干个管家基因同时进行表达量分析,从中选择表达量变化幅度小的管家基因来使用,选择最适校正基因的软件有很多,请下载利用(geNorm、BestKeeper等)。
【参考文献】
Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Vandesompele
J,(2002)3(7): RESEARCH0034.1-11
为了保持与实际检测样品间的扩增效率的一致性,作为标准品应尽量选择与实际检测样品结构近似的样品。例如以基因组DNA为起始材料时就要选择基因组DNA作为标准品,进行mRNA表达解析时最好选择表达目的基因的Total RNA(或以Total RNA为模板合成的cDNA)作为标准品。即使扩增碱基序列相同,但整体模板不同,也有可能导致PCR扩增效率不同(比如基因组DNA和质粒DNA等)。
Real Time PCR制作标准曲线有2种方法:(1) 从RNA开始稀释进行反转录和Real Time PCR的标准曲线制作;
(2) 反转录反应后得到的cDNA开始稀释进行Real Time PCR的标准曲线制作。根据实验目的不同,选择合适的方法。
[绝对定量]
RNA绝对定量要考虑到反转录效率,应选择上述方法(1)的标准品。此时,不推荐使用cDNA稀释品作为标准品。
[相对定量]
相对定量通常使用管家基因等来测定,可以校正反转录效率的误差,推荐选择上述方法(2)的标准品。如果使用RNA制作标准曲线,除了PCR扩增效率以外,还要考虑RNA的反转录效率。如果按此与实际情况不符的PCR扩增效率计算,可能会导致计算结果有偏差。
1. 引物设计时避免基因组DNA扩增:首选确认目的基因的基因组结构,选择跨越较长的内含子。然后在这个内含子两侧的外显子上分别设计上、下游引物。
2. 使用DNase I处理去除基因组DNA:使用常规方法提取Total RNA后,再使用DNase I分解混入的基因组DNA。
3. 使用RR047A PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) 附带的gDNA Eraser可以高效去除残留的基因组DNA。仅需42℃、2分钟即可去除基因组DNA。后续反转录反应需15分钟。全程反应不超过20分钟。
使用验证过的引物时,之前的融解曲线分析(Tm值)可作为参考依据。如果Tm值与过去的验证结果相同,可以认为qPCR扩增产物与过去验证结果相同。
但是,请切记,相同的PCR产物具有相同的Tm值,但仅凭相同的Tm值并不一定可以确认PCR产物是相同的。应通过另外的方法来确认。如果是首次使用该引物,应使用电泳方法确认qPCR产物是否是预期的大小。
qPCR的灵敏度取决于实验条件,如使用的试剂和引物。已经证实合适的反应系统可以检测低至10个拷贝的模板。
1. 交点法(Crossing point法),是通过阈值和扩增曲线的交点计算Ct值的方法。
2. 二次求导法(2nd Derivative Maximum法),是通过扩增曲线二次求导后取其最大的点计算Ct值的方法。
后一种方法是以扩增速度的变化率最大点计算Ct值,不会随阈值的设定不同而变化,所以重现性高。另外,还能排除仪器检出误差的影响,是一种高准确度的计算方法。但是,并不是所有厂家的定量PCR仪器都具有二次求导计算Ct值的功能。
页面更新:2020-04-24 13:57:27


 京公网安备 110114000405号
京公网安备 110114000405号